La hemofilia es el resultado de mutaciones en los loci del factor VIII o IX en el cromosoma X y cada una se presenta en formas leves, moderadas y graves.
Contenidos
¿Qué es la hemofilia?
La hemofilia ahora está aumentando entre la población pediátrica de todo el mundo.
- La hemofilia es el resultado de mutaciones en los loci del factor VIII o IX en el cromosoma X y cada una se presenta en formas leves, moderadas y graves.
- Un nivel similar de deficiencia de factor VIII o IX da como resultado una enfermedad clínicamente indistinguible porque el resultado final es una activación deficiente del factor X por el complejo de factor Xasa (FVIIIa / FIXa / calcio y fosfolípido).
- La hemofilia A es un trastorno recesivo ligado al cromosoma X causado por la deficiencia del factor VIII de coagulación plasmática funcional (FVIII), que puede heredarse o surgir de una mutación espontánea.
- La hemofilia B, o enfermedad de Christmas , es un trastorno recesivo hereditario ligado al cromosoma X que provoca la deficiencia del factor IX de coagulación plasmática funcional.
Fisiopatología
Las fisiopatologías de la hemofilia A y B son las siguientes:

Hemofilia A
- Se cree que los sitios primarios de producción de factor VIII (FVIII) son el endotelio vascular en el hígado y el sistema reticuloendotelial.
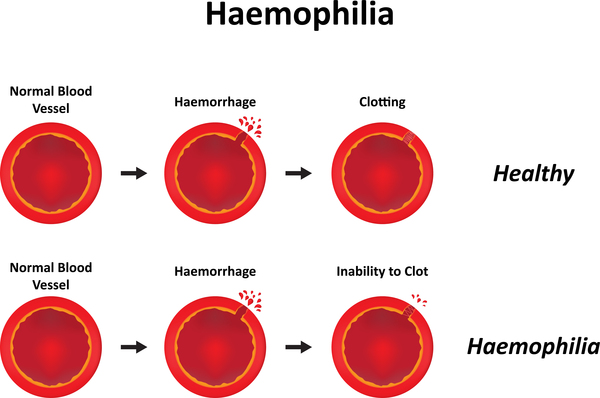
- La deficiencia de FVIII, el FVIII disfuncional o los inhibidores de FVIII conducen a la interrupción de la cascada de coagulación intrínseca normal, lo que da como resultado una hemorragia excesiva en respuesta a un traumatismo y, en casos graves, una hemorragia espontánea.
- Las células sinoviales humanas sintetizan altos niveles de inhibidor de la vía del factor tisular, lo que da como resultado un mayor grado de inhibición del factor Xa (FXa), que predispone a las articulaciones hemofílicas a sangrar.
- Este efecto también puede explicar la respuesta espectacular de las infusiones de factor VII activado (FVIIa) en pacientes con hemartrosis aguda e inhibidores de FVIII.
- El sangrado en una articulación puede provocar inflamación sinovial, que predispone a la articulación a más hemorragias; una articulación que ha tenido hemorragias repetidas (según una definición, al menos 4 hemorragias en un período de 6 meses) se denomina articulación objetivo.
- Aproximadamente el 30% de los pacientes con hemofilia A grave desarrollan inhibidores de aloanticuerpos que pueden unirse al FVIII; estos inhibidores son típicamente inmunoglobulina G (IgG), predominantemente de la subclase IgG4, que neutraliza los efectos coagulantes de la terapia de reemplazo.

Hemofilia B
- La deficiencia de factor IX, el factor IX disfuncional o los inhibidores del factor IX conducen a la interrupción de la cascada de coagulación intrínseca normal, lo que resulta en hemorragia espontánea y / o hemorragia excesiva en respuesta al trauma.
- Los sitios de hemorragia incluyen articulaciones (p. Ej., Rodilla, codo), músculos, sistema nervioso central (SNC), sistema GI, sistema genitourinario (GU), sistema pulmonar y sistema cardiovascular .
- El factor IX, una glicoproteína monocatenaria dependiente de la vitamina K, es sintetizada primero por el hepatocito; la proteína precursora sufre una extensa modificación postraduccional antes de ser secretada a la sangre.
- El sistema intrínseco se inicia cuando el factor XII se activa por contacto con el endotelio dañado.
- En el sistema extrínseco, la conversión del factor X en factor Xa implica factor tisular (TF) o tromboplastina; factor VII; e iones de calcio.
- FVIII y FIX circulan en forma inactiva; cuando se activan, estos 2 factores cooperan para escindir y activar el factor X, una enzima clave que controla la conversión de fibrinógeno en fibrina.
- Por lo tanto, la falta de cualquiera de estos factores puede alterar significativamente la formación de coágulos y, como consecuencia, provocar una hemorragia clínica.
Estadísticas e incidencias
La hemofilia está progresando lentamente entre los pacientes pediátricos en todas partes del mundo.
- La hemofilia A es la enfermedad genética ligada al cromosoma X más común y la segunda deficiencia de factor más común después de la enfermedad de von Willebrand (EvW).
- La incidencia mundial de hemofilia A es de aproximadamente 1 caso por cada 5000 hombres, y aproximadamente un tercio de las personas afectadas no tienen antecedentes familiares del trastorno.
- En los Estados Unidos, la prevalencia de hemofilia A es de 20,6 casos por 100.000 hombres; En 2016, se estimó que la cantidad de personas con hemofilia en los Estados Unidos era de unas 20.000.
- La hemofilia A se presenta en todas las razas y grupos étnicos.
- Dado que la hemofilia es una afección recesiva ligada al cromosoma X, se presenta predominantemente en hombres; las mujeres suelen ser portadoras asintomáticas.
- Se estima que la incidencia de hemofilia B es de aproximadamente 1 caso por cada 25.000-30.000 nacimientos de varones.
- La prevalencia de hemofilia B es de 5,3 casos por cada 100.000 varones, y el 44% de los que padecen una enfermedad grave.
- La hemofilia B es mucho menos común que la hemofilia A. De todos los casos de hemofilia, 80-85% son hemofilia A, 14% son hemofilia B y el resto son varias otras anomalías de la coagulación.
- La hemofilia B ocurre en todas las razas y grupos étnicos.
Causas
Las causas de la hemofilia A y B son aparentemente de una forma genética.
- Genética. La hemofilia A es causada por una mutación genética heredada o adquirida que resulta en una disfunción o deficiencia del factor VIII, o por un inhibidor adquirido que se une al factor VIII; La hemofilia B es una enfermedad recesiva ligada al cromosoma X causada por una mutación heredada o adquirida en el gen del factor IX o por un inhibidor del factor IX adquirido.
Manifestaciones clínicas
La hemofilia es sugerida por antecedentes de hemorragia desproporcionada al trauma o de hemorragia espontánea, o antecedentes familiares de problemas hemorrágicos.
- Hemorragia espontánea. Aproximadamente el 30-50% de los pacientes con hemofilia grave presentan manifestaciones de hemorragia neonatal (p. Ej., Después de la circuncisión); otros recién nacidos pueden presentar un hematoma severo y sangrado prolongado del cordón o el área umbilical o en los sitios de extracción de sangre o inmunizaciones.
- Hematuria . En el tracto genitourinario, la hematuria macroscópica puede ocurrir hasta en el 90% de los pacientes.
- Síntomas generales. Puede ocurrir debilidad y ortostasis.
- Musculoesquelético. El hormigueo, el crujido, el calor, el dolor, la rigidez y la negativa a usar la articulación entre los niños son comunes.
- Sistema nervioso central. Puede causar dolor de cabeza, rigidez en el cuello, vómitos , letargo, irritabilidad y síndromes de la médula espinal .
- Genitourinario. Los síntomas pueden ser indoloros; puede haber dolor hepático / esplénico y signos peritoneales.
Hallazgos de evaluación y diagnóstico
El diagnóstico de hemofilia A y B se establece midiendo lo siguiente:
- Ensayo cromogénico. Algunos consideran que este ensayo es más preciso, ya que mide el nivel de actividad del factor VIII en plasma, pero está menos disponible en los laboratorios clínicos de los Estados Unidos.
- Estudios de laboratorio. Los estudios de laboratorio para la sospecha de hemofilia incluyen un hemograma completo, estudios de coagulación y un análisis de factor VIII (FVIII).
- Tomografías computarizadas. Las tomografías computarizadas de la cabeza sin contraste se utilizan para evaluar la hemorragia intracraneal espontánea o traumática.
- Resonancia magnética. Realizar imágenes de resonancia magnética (IRM) en la cabeza y la columna vertebral para una evaluación adicional de hemorragia espontánea o traumática; La resonancia magnética también es útil en la evaluación del cartílago, la membrana sinovial y el espacio articular.
- Ecografía. La ecografía es útil en la evaluación de articulaciones afectadas por derrames agudos o crónicos.
- Prueba de inhibidores. La confirmación de laboratorio de un inhibidor del FVIII es clínicamente importante cuando un episodio hemorrágico no se controla a pesar de la infusión de cantidades adecuadas de concentrado de factor.
- Prueba de portador. El cribado del estado de portador se puede realizar midiendo la relación entre la actividad coagulante del FVIII y la concentración del antígeno del factor von Willebrand (vWF); una proporción inferior a 0,7 sugiere la condición de portador.
- Radiografía. La radiografía para la evaluación de las articulaciones tiene un valor limitado en la hemartrosis aguda; La evidencia de enfermedad articular degenerativa crónica puede ser visible en las radiografías en pacientes que no han sido tratados o tratados inadecuadamente o en aquellos con hemorragias articulares recurrentes.
Administración medica
El tratamiento de la hemofilia puede incluir profilaxis, manejo de episodios hemorrágicos, tratamiento de inhibidores del factor VIII (FVIII) y tratamiento y rehabilitación de la sinovitis por hemofilia.
- Atención prehospitalaria. El transporte rápido a la atención definitiva es el pilar de la atención prehospitalaria; Los proveedores de atención prehospitalaria deben aplicar técnicas hemostáticas agresivas, ayudar a los pacientes capaces de autoadministrarse la terapia con factor y recopilar datos históricos específicos si el paciente no puede comunicarse.
- Atención en el departamento de emergencias. Utilice técnicas hemostáticas agresivas; corregir la coagulopatía de inmediato; incluir un estudio de diagnóstico para la hemorragia, pero nunca retrasar la corrección de la coagulación indicada en espera de las pruebas de diagnóstico; sangrado articular agudo y hematomas grandes en expansión que requieren un reemplazo de factor adecuado durante un período prolongado hasta que el sangrado comienza a resolverse, como lo demuestran los métodos clínicos y / o objetivos; Los episodios hemorrágicos potencialmente mortales generalmente se tratan inicialmente con niveles de FVIII de aproximadamente el 100%, hasta que la situación clínica justifique una reducción gradual de la dosis .
- Concentrados de factor VIII y FIX. Se encuentran disponibles varios concentrados de FVIII y FIX para tratar la hemofilia A y B; además de mejorar la hemostasia, la infusión continua disminuye la cantidad de factor utilizado, lo que puede resultar en ahorros significativos; Obtenga análisis de nivel de factor diariamente antes de cada infusión para establecer un patrón estable de reemplazo con respecto a la dosis y frecuencia de administración.
- Desmopresina . El análogo de desmopresina vasopresina, o 1-desamino-8-D-arginina vasopresina (DDAVP), se considera el tratamiento de elección para la hemofilia A leve y moderada; La DDAVP estimula un aumento transitorio de los niveles plasmáticos de FVIII; La DDAVP puede dar como resultado una hemostasia suficiente para detener un episodio hemorrágico o para preparar a los pacientes para procedimientos dentales y quirúrgicos menores.
- Manejo del sangrado. La inmovilización de la extremidad afectada y la aplicación de bolsas de hielo son útiles para disminuir la hinchazón y el dolor; La infusión temprana tras el reconocimiento de los síntomas iniciales de una hemorragia articular a menudo puede eliminar la necesidad de una segunda infusión al prevenir la reacción inflamatoria en la articulación; La terapia de reemplazo rápida y adecuada es la clave para prevenir complicaciones a largo plazo.
- Tratamiento de pacientes con inhibidores. Los inhibidores son anticuerpos que neutralizan el factor VIII (FVIII) y pueden hacer que la terapia de reemplazo sea ineficaz; el tratamiento de pacientes con inhibidores de FVIII es difícil; asumiendo que no hay respuesta anamnésica, los inhibidores de títulos bajos (es decir, concentraciones por debajo de 5 unidades Bethesda [BU]) ocasionalmente pueden superarse con dosis altas de factor VIII; No existe un tratamiento establecido para los episodios hemorrágicos en pacientes con inhibidores de títulos altos.
- Infusiones profilácticas de factor. El objetivo principal del tratamiento profiláctico es prevenir los síntomas hemorrágicos y el daño de los órganos, en particular de las articulaciones; en diciembre de 2013, la Administración de Drogas y Alimentos de los EE. UU . (FDA) amplió la indicación del complejo coagulante antiinhibidor (Feiba NF) para incluir la profilaxis de rutina en pacientes con hemofilia A o B que han desarrollado inhibidores; la aprobación se basó en los datos de un estudio fundamental de fase III en el que un régimen profiláctico resultó en una reducción del 72% en la tasa media anual de hemorragias en comparación con el tratamiento a demanda.
- El manejo del dolor. La artropatía crónica hemofílica se asocia con dolor; Se han utilizado agentes narcóticos, pero el uso frecuente de estas drogas puede resultar en adicción; En su lugar, se pueden utilizar fármacos antiinflamatorios no esteroideos porque sus efectos sobre la función plaquetaria son reversibles y porque estos fármacos pueden ser eficaces en el tratamiento del dolor artrítico agudo y crónico; Evite la aspirina debido a su efecto irreversible sobre la función plaquetaria.
- Actividad. Generalmente, las personas con hemofilia severa deben evitar los deportes de contacto de alto impacto y otras actividades con un riesgo significativo de trauma; sin embargo, cada vez hay más pruebas que sugieren que la actividad física adecuada mejora el acondicionamiento general, reduce la tasa y la gravedad de las lesiones y mejora el funcionamiento psicosocial.
- Terapia de genes. Con la clonación del FVIII y los avances en tecnologías moleculares, se concibió la posibilidad de curar la hemofilia con terapia génica; terapia génica ex vivo, en la que las células a trasplantar se modifican genéticamente para secretar factor VIII y luego se reimplantan en el receptor; terapia génica in vivo, en la que un vector (típicamente un virus alterado para incluir ADN de FVIII) se inyecta directamente en el paciente; y terapia génica no autóloga, en la que las células modificadas para secretar FVIII se empaquetan en dispositivos inmunoprotegidos y se implantan en receptores.
- Radiosinovectomía. En pacientes que desarrollan sinovitis por hemorragias articulares, la inyección intraarticular de radioisótopos para extirpar la membrana sinovial (radiosinovectomía) se puede utilizar para disminuir el sangrado, retrasar la progresión del daño cartilaginoso y óseo y prevenir la artropatía.
Manejo farmacológico
Los medicamentos de elección para pacientes con hemofilia son:
- Factor VIII. El factor VIII (FVIII) es el tratamiento de elección para la hemorragia aguda o potencial en la hemofilia A; el concentrado de FVIII recombinante es generalmente la fuente preferida de factor VIII; La administración profiláctica de FVIII a menudo se recomienda para pacientes pediátricos con enfermedad grave.
- Agentes antifibrinolíticos. Los antifibrinolíticos, como el ácido aminocaproico y el ácido tranexámico, son especialmente útiles para las hemorragias de la mucosa oral, pero están contraindicados como tratamientos iniciales para la hematuria relacionada con la hemofilia que se origina en el tracto urinario superior porque pueden causar uropatía obstructiva o anuria.
- Factor IX. El factor IX es el tratamiento de elección para la hemorragia aguda o la supuesta hemorragia aguda en la hemofilia B. El factor IX recombinante es la fuente preferida para la terapia de reemplazo.
- Factor de coagulación VIIa. Estos agentes pueden activar el factor de coagulación X a factor Xa, así como el factor de coagulación IX a IXa.
- Factores de coagulación. Los concentrados de FVIII reemplazan el FVIII deficiente en pacientes con hemofilia A, con el objetivo de lograr una respuesta hematológica normal a la hemorragia o prevenir la hemorragia; los productos recombinantes deben usarse inicialmente y posteriormente en todos los casos de hemofilia recién diagnosticados que requieran reemplazo de factor; Los agentes que evitan la actividad del FVIII en la cascada de la coagulación (p. ej., FVII activado) se utilizan en pacientes con inhibidores de FVIII.
- Agentes antihemofílicos. Estos agentes se utilizan para controlar el sangrado en la hemofilia B o la deficiencia de FIX y para prevenir y / o controlar el sangrado en pacientes con hemofilia A e inhibidores del FVIII.
- Anticuerpos monoclonicos. Los anticuerpos monoclonales se usan para unirse a una sustancia específica en el cuerpo (por ejemplo, moléculas, antígenos); esta unión es muy versátil y puede imitar, bloquear o provocar cambios para ejecutar mecanismos precisos (p. ej., moléculas puente, sustitución o activación de enzimas o cofactores, estimulación del sistema inmunológico).
- Relacionados con la vasopresina. La desmopresina aumenta transitoriamente el nivel plasmático de FVIII en pacientes con hemofilia A leve.
La gestión de enfermería
La atención de enfermería para un niño con hemofilia incluye:
Evaluación de enfermería
La evaluación en un niño con hemofilia incluye lo siguiente:
- Historia. En el caso de pacientes en los que se sospeche hemofilia, pregunte acerca de antecedentes de hemorragia desproporcionada al trauma, antecedentes de hemorragia espontánea, trastornos hemorrágicos en la familia y enfermedades concomitantes (especialmente aquellas asociadas con hemofilia adquirida, como trastornos inflamatorios crónicos, enfermedades autoinmunes, enfermedades hematológicas). neoplasias malignas y reacciones alérgicas a medicamentos).
- Examen físico. Evaluar la inflamación de las articulaciones y la capacidad para mover la extremidad afectada; Valorar la amplitud de movimiento limitada, las contracturas y los cambios óseos en las articulaciones cuando se haya detenido la hemorragia.
Diagnósticos de enfermería
Según los datos de la evaluación, los principales diagnósticos de enfermería son:
- Dolor agudo relacionado con una lesión traumática de los músculos.
- Dolor y malestar relacionados con lesiones físicas deterioradas con la aparición de episodios hemorrágicos.
- Afrontamiento familiar comprometido relacionado con información o comprensión incorrecta e inadecuada.
- Riesgo de hemorragia relacionado con la disminución de la concentración de factores de coagulación que circulan en la sangre (factor VIII y factor IX).
- Riesgo de lesión relacionada con la disminución del factor de coagulación (VIII o IX).
Planificación y objetivos de la atención de enfermería
Artículo principal 5 Planes de atención de enfermería de hemofilia
Los principales objetivos son:
- El niño experimentará una disminución del dolor.
- El niño mantendrá una movilidad física óptima como lo demuestra el rango de movimiento normal (ROM) y las actividades de la vida diaria dentro de sus posibilidades.
- La familia se enfrentará eficazmente a la enfermedad del niño.
- El riesgo del niño de sufrir lesiones por posible sangrado se reduce mediante el uso de medidas profilácticas adecuadas.
Intervenciones de enfermería
Las intervenciones de enfermería para un niño con hemofilia son:
- Aliviar el dolor. Inmovilice las articulaciones y aplique vendajes elásticos a la articulación afectada si está indicado; eleve el afectado y aplique una compresa fría en los sitios sangrantes activos, pero debe usarse con precaución en niños pequeños para evitar la rotura de la piel.
- Mantener una movilidad física óptima. Proporcione un ejercicio de ROM suave y pasivo cuando la condición del niño sea estable; educar sobre medidas preventivas, como la aplicación de equipos de protección y la administración de productos de factor; y derivar a consultas de fisioterapia, terapia ocupacional y ortopédica, según sea necesario.
- Ayudar en el afrontamiento de la familia. Anime a los miembros de la familia a verbalizar las áreas problemáticas y a desarrollar soluciones por su cuenta; Anime a los miembros de la familia a expresar sus
sentimientos, por ejemplo, cómo tratan las necesidades crónicas de un miembro de la familia y los patrones de afrontamiento que ayudan o dificultan la adaptación a los problemas. - Evita el sangrado. Controle los niveles de hemoglobina y hematocrito; evaluar el anticuerpo inhibidor del factor VIII; anticipar o instruir sobre la necesidad de tratamiento profiláctico ante situaciones de alto riesgo, como procedimientos quirúrgicos o diagnósticos invasivos, o trabajos dentales; y proporcionar terapia de reemplazo de factores de coagulación deficientes.
- Prevenga lesiones. Utilice juguetes apropiados (blandos, no puntiagudos
o pequeños objetos afilados); en el caso de los bebés, es posible que sea necesario utilizar barandas de cama acolchadas en la cuna; evitar la temperatura rectal; proporcionar una higiene bucal adecuada (uso de
un dispositivo de irrigación de agua; uso de un cepillo de dientes suave o ablandamiento del cepillo de dientes con agua tibia antes de cepillarse; uso de un cepillo de dientes con punta de esponja); y evite los deportes de contacto como fútbol, fútbol, hockey sobre hielo, kárate.
Evaluación
Las metas se alcanzan como lo demuestra:
- El niño experimentó una disminución del dolor.
- El niño mantuvo una movilidad física óptima como lo demuestra el rango de movimiento normal (ROM) y las actividades de la vida diaria dentro de sus capacidades.
- La familia se enfrentó eficazmente a la enfermedad del niño.
- El riesgo del niño de sufrir lesiones por posible hemorragia se redujo mediante el uso de medidas profilácticas adecuadas.
Pautas de documentación
La documentación en un niño con hemofilia incluye lo siguiente:
- Resultados de la evaluación inicial y posterior para incluir signos y síntomas.
- Restricciones culturales o religiosas individuales y preferencias personales.
- Plan de cuidados y personas involucradas.
- Plan de enseñanza.
- Respuestas del paciente a las enseñanzas, intervenciones y acciones realizadas.
- Logro o progreso hacia el resultado deseado.
- Necesidades a largo plazo y quién es responsable de las acciones a tomar.
Prueba de práctica: Hemofilia
Aquí hay un cuestionario de 5 elementos para la guía de estudio de la hemofilia. Visite nuestra página del banco de pruebas de enfermería para obtener más preguntas sobre la práctica de NCLEX .
1. El hijo del Sr. y la Sra. Smith tiene hemofilia; ¿Cuál de las siguientes acciones les indicaría que evitaran?
A. Bajando el área lesionada.
B. Inmovilización de la articulación.
C. Aplicar presión.
D. Aplicar frío en la zona.
1. Respuesta: A. Bajar el área lesionada.
- Opción A: con hemofilia, el área lesionada debe elevarse, no bajarse.
- Opciones B, C y D: inmovilizar la articulación y aplicar frío o presión en el área son medidas adecuadas para controlar el sangrado.
2. ¿Cuál de las siguientes pruebas de laboratorio es más eficaz para diagnosticar la hemofilia?
A. Conteo sanguíneo completo (CBC).
B. Tiempo de sangrado (BT).
C. Recuento de plaquetas (PC).
D. Tiempo de tromboplastina parcial (PTT).
2. Respuesta: D. Tiempo parcial de tromboplastina (PTT).
- Opción D: PTT es anormal en la hemofilia. Por lo tanto, esta prueba será la más útil para diagnosticar el trastorno.
- Opción A: el CBC no se ve afectado en la hemofilia.
- Opciones B y C: el tiempo de sangrado y el recuento de plaquetas son normales en la hemofilia.
3. Un niño con hemofilia A conocida fue llevado a la sala de emergencias con quejas de hemorragia nasal y algunos hematomas en las articulaciones. ¿Cuál de los siguientes debe anticipar la enfermera que se le dará al niño?
A. Ciclosporina .
B. Suplemento oral de hierro.
C. Factor VIII.
D. Factor X.
3. Respuesta: C. Factor VIII.
- Opción C: La hemofilia A, también llamada deficiencia del factor VIII (FVIII) o hemofilia clásica, es un trastorno genético causado por la falta o defecto del factor VIII, una proteína de la coagulación. El tratamiento inicial es la administración de factor VIII para reemplazar el factor faltante y disminuir el episodio hemorrágico.
- Opciones A, B, D : Estos medicamentos no se utilizan en este caso.
4. La madre de un niño con hemofilia pregunta a la enfermera qué medicamento de venta libre es adecuado para las molestias articulares de su hijo. La enfermera debe decirle a la madre que compre:
A. Aspirina (ácido acetilsalicílico).
B. Naproxeno (Naprosyn).
C. Tylenol (acetaminofén).
D. Advil ( ibuprofeno ).
4. Respuesta: C. Tylenol (acetaminofén).
- Opción C: La enfermera debe recomendar acetaminofén para las molestias articulares del niño porque no afectará el tiempo de sangrado.
- Opciones A, B, D: Las respuestas A, C y D son medicamentos antiinflamatorios no esteroides que pueden prolongar el tiempo de sangrado; por lo tanto, no son adecuados para niños con hemofilia.
5. ¿Cuál de los siguientes trastornos resulta de una deficiencia de factor VIII?
A. Enfermedad de células falciformes.
B. Enfermedad de Christmas.
C. Hemofilia A.
D. Hemofilia B.
5. Respuesta: C. Hemofilia A.
- Opción C: la hemofilia A es el resultado de una deficiencia de factor VIII.
- Opción A: la anemia de células falciformes es causada por una molécula de hemoglobina defectuosa.
- Opciones B y D: la enfermedad de Christmas, también llamada hemofilia B, produce una deficiencia de factor IX.
Ver también
Temas relacionados con esta guía de estudio:
- Guías de estudio de enfermería pediátrica
- Notas de enfermería: guías de estudio para varios temas
- Preguntas de práctica de NCLEX de enfermería pediátrica
Otras lecturas
Recursos y libros recomendados para enfermería pediátrica:
- PedsNotes: Guía clínica de bolsillo para enfermeras (Guías clínicas de bolsillo para enfermeras)
- La enfermería pediátrica es increíblemente fácil
- Fundamentos de enfermería pediátrica de Wong
- Enfermería pediátrica: los componentes críticos del cuidado de enfermería